Pathways Case Record: A Patient With Cirrhosis and Small Bowel Edema
In This Case Study
- A 39-year-old man presented with an acute episode of severe epigastric pain
- The patient had a notable history of decompensated cirrhosis of unknown etiology complicated by ascites, spontaneous bacterial peritonitis, and esophageal varices
- Initial evaluation revealed extensive small bowel edema and C1 esterase inhibitor (C1-INH) deficiency. His presentation was most consistent with angioedema localized in the small bowel
- The Pathways Consult Service was consulted to investigate the pathophysiologic mechanism for his dramatic small bowel edema
- The team focused on two primary questions: (1) Does liver dysfunction predispose to angioedema, and (2) why did the patient present with abdominal pain during his episode?
A 39-year-old man presented with an acute episode of severe epigastric pain. The patient had a notable history of decompensated cirrhosis of unknown etiology complicated by ascites, spontaneous bacterial peritonitis, and esophageal varices. Additionally, he was post-right hemiglossectomy and chemoradiation for a squamous cell carcinoma on his tongue. Initial evaluation revealed extensive small bowel edema and C1 esterase inhibitor (C1-INH) deficiency. His presentation was most consistent with angioedema localized in the small bowel. Following treatment with high doses of antihistamines and intermittent opioids, the patient was discharged.
Subscribe to the latest updates from Advances in Motion
The Pathways Consult Service in the Department of Medicine at Massachusetts General Hospital investigated the pathophysiologic mechanism for his dramatic small bowel edema. The Pathways team focused on two primary questions:
- Does liver dysfunction predispose to angioedema?
- Why did the patient present with abdominal pain during his episode?
Background and Diagnosis
Angioedema is self-limited, localized subcutaneous or submucosal swelling caused by fluid accumulation in interstitial tissues. Affected areas typically include regions with loose connective tissue such as the face, mouth, extremities, genitalia, and bowel walls. Angioedema is divided broadly into either mast cell-mediated (histaminergic) or bradykinin-mediated angioedema (Int J Emerg Med). Allergens triggered mast cell-mediated angioedema (e.g., food) and were characterized by signs of mast cell degranulation presenting as hives, itchy skin, flushed skin, and bronchospasms. On the contrary, bradykinin-mediated angioedema lacks these allergic symptoms. The peptide bradykinin, which promotes inflammation, is regulated by an interaction between the kallikrein-kinin, renin-angiotensin, and complement pathways (Int Arch Allergy Immunol) (Figure 1). Bradykinin binds to the bradykinin B2 receptor leading to vasodilation and angioedema. The glycoprotein C1-INH regulates this action by inhibiting factor XII and kallikrein (Immunobiology). Additionally, angiotensin-converting enzyme in the renin-angiotensin system degrades bradykinin into inactive products (Int J Emerg Med).
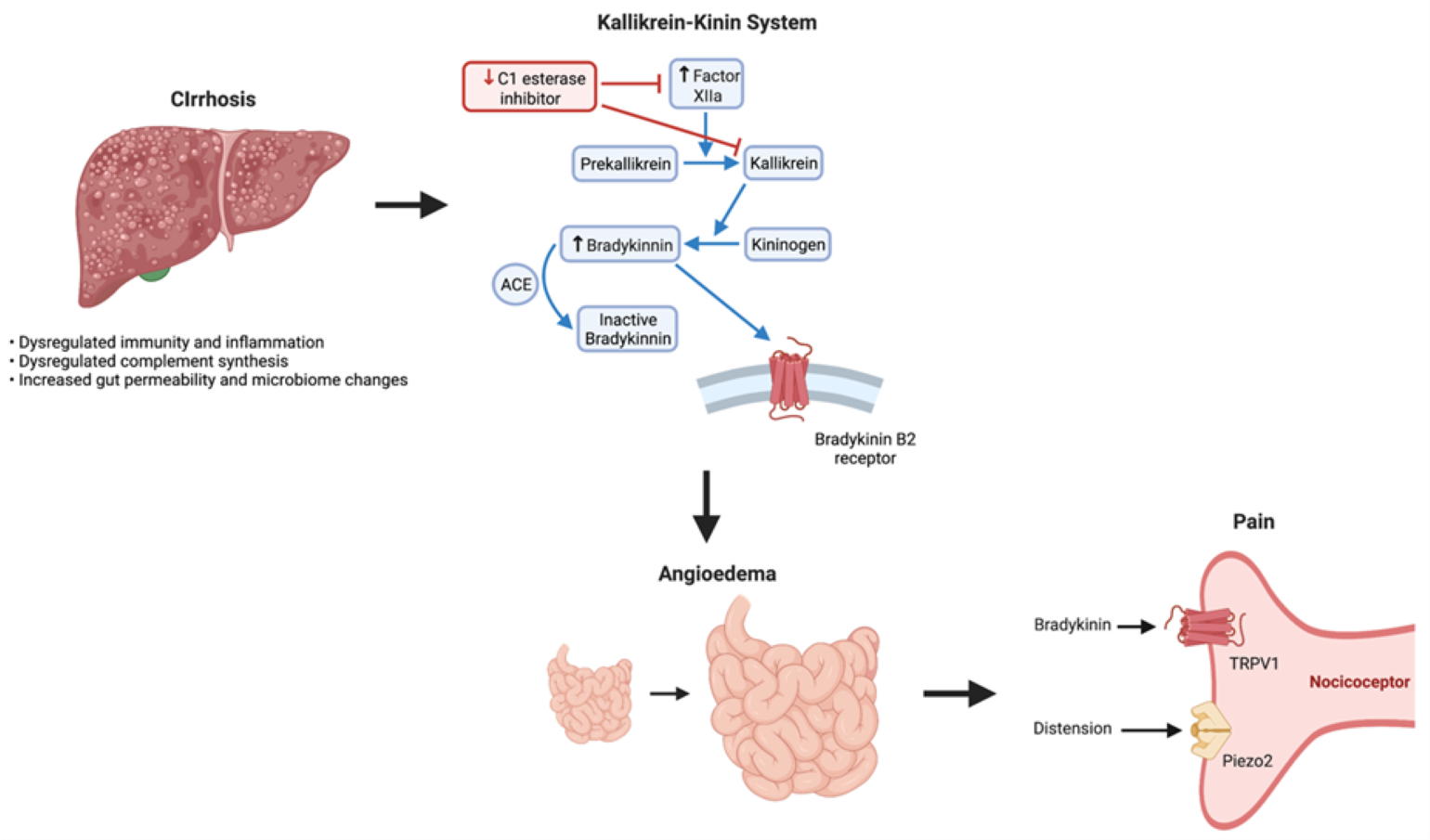
Figure 1
Kallikrein-Kinin System.
Bradykinin-mediated angioedema can be further classified as idiopathic, acquired, or hereditary (N Engl J Med). Acquired forms are often attributed to medications (e.g., ACE-inhibitors, non-steroidal anti-inflammatory drugs) or C1-INH deficiency. C1-INH deficiency may be caused by increased C1-INH consumption with complement pathway dysregulation, decreased C1-INH production due to liver disease, or C1-INH autoantibodies from autoimmune or lymphoproliferative disorders. Hereditary angioedema (HAE) stems from either an impaired production of C1-INH with reduced levels and function (type I) or C1-INH produced is dysfunctional (type II) (Postgrad Med). Both HAE subtypes are caused by mutations in the SERPING1 gene, which encodes the C1-INH peptide. Alternatively, HAE may also occur due to gene mutations related to other steps in the bradykinin pathway, such as factor XII (F12).
The main functional cells in the liver, hepatocytes, are the primary source of C1-INH. Although liver disease has not been extensively investigated as a trigger for angioedema, it has been suggested that HAE may be a metabolic liver disorder rather than an isolated enzyme deficiency. Indeed, some reports correlate HAE with an increased risk of nonalcoholic fatty liver disease. Did our patient's underlying cirrhosis contribute to his angioedema episode? If so, by what mechanism could this occur? Cirrhosis leads to a persistent pro-inflammatory state through increased inflammatory pathway activation, cytokine production, and levels of endothelial activation molecules (e.g., vascular endothelial growth factor [VEGF]). Intriguingly, circulating levels of VEGF and other vascular permeability factors are elevated in HAE patients and correlate with disease severity. VEGF and bradykinin act synergistically to promote vascular leak, edema, and pain in angioedema. In addition to the inflammatory state, it is possible that autoantibodies against C1-INH (present in 12% of cirrhosis patients) may account for the development of angioedema (Ann Intern Med). Lastly, patients with alcohol-related cirrhosis have altered microbiota, including a shift towards pathogenic bacteria and increased risk for local and systemic translocation (World J Gastroenterol, Nat Rev Gastroenterol Hepatol). If present in our patient with decompensated cirrhosis, this may explain the local edema.
The Pathways team next focused on the mechanism of abdominal pain in our patient. Nociceptors are specialized sensory neurons that detect thermal, mechanical, and chemical stimuli from both the internal and external environment to mediate pain (J Clin Invest). For example, in the setting of infection and tissue injury, immune cells release inflammatory cytokines that directly activate nociceptors (Trends Immunol). Furthermore, these neurons express pattern recognition receptors that recognize components of pathogens (i.e., pathogen-associated molecular patterns) and endogenous danger signals (i.e., danger-associated molecular patterns). Nociceptive neurons densely innervate peripheral tissues, including skin, lungs, and gastrointestinal tract. In our patient, mechano- and chemo-sensitive nociceptors likely contribute to the abdominal pain pathophysiology. The expression of nociceptors of two ion channels, Piezo2 (a mechanosensitive ion channel involved in proprioception) and TRPV1 (an ion channel that detects heat and noxious chemicals including bradykinin), are prime candidates for mediating our patient's abdominal pain in the setting of striking submucosal bowel edema (Trends Biochem Sci, Nat Rev Drug Discov).
The release of neuropeptides following nociceptor activation may provide an alternate mechanism driving the pain in this patient. These neuropeptides exert potent effects on the vasculature and immune cells to regulate inflammation (Trends Immunol). For example, calcitonin gene-related peptide (CGRP) released by nociceptors activates receptors on vascular smooth cell cells to promote muscle relaxation and vasodilation. Substance P activates receptors on vascular endothelial cells to increase vascular permeability. CGRP and substance P also act on lymphatic endothelial cells to regulate lymph flow. Taken together, these neuropeptides can drive the formation of edema.
Summary and Future Steps
We propose that our patient presenting with acute abdominal pain and small bowel edema in the setting of possible angioedema was mediated by reduction of C1-INH triggered by liver disease, with his pain driven by sensory neural pathways (Figure 1). While cirrhosis has not been extensively studied in the context of angioedema, it is possible that it contributed to clinical presentation in our patient. Utilizing new culture methods from liver biopsies (JHEP Rep) may provide a window into the contribution of liver disease in HAE. Furthermore, direct testing for C1-INH autoantibodies in our patient would be beneficial. Additional studies investigating pan-neuronal markers in intestinal biopsies to evaluate the density of innervation and serum levels of neuroactive agents (e.g., CGRP and substance P) may provide insights into his abdominal pain.
Learn about the Pathways Consult Service at Mass General
Explore research in the Department of Medicine