Pathways Case Record: Familial Dilated Cardiomyopathy
In This Case Study
- A 29-year-old woman with a family history notable for a fraternal twin sister with dilated cardiomyopathy requiring a heart transplant presented with two months of fatigue, joint pain, non-productive cough, and intermittent fevers
- She was found to have acute decompensated heart failure due to newly diagnosed dilated cardiomyopathy (DCM)
- Targeted genetic testing was performed, and she was found to have a likely pathogenic heterozygous mutation in the TTN gene (c.49648+2del)
- The Pathways Service at Massachusetts General Hospital was consulted and focused on characterizing the likely impact of TTN mutation on cardiomyocyte function and its potential causative role in DCM
A 29-year-old woman with a family history notable for a fraternal twin sister with dilated cardiomyopathy requiring a heart transplant presented with two months of fatigue, joint pain, non-productive cough, and intermittent fevers. She developed progressive dyspnea two weeks prior to presentation. She was found to have acute decompensated heart failure due to newly diagnosed dilated cardiomyopathy (DCM). She developed cardiogenic shock requiring inotropic support and ultimately underwent left ventricular assist device (LVAD) placement as a bridge to a potential heart transplant. An extensive workup for ischemic, infectious, autoimmune, nutritional, and infiltrative causes of DCM was ultimately unremarkable. Therefore, targeted genetic testing was performed, and she was found to have a likely pathogenic heterozygous mutation in the TTN gene (c.49648+2del).
Subscribe to the latest updates from Advances in Motion
The Pathways Service in the Department of Medicine at Massachusetts General Hospital was consulted and focused on characterizing the likely impact of TTN mutation on cardiomyocyte function and its potential causative role in DCM, driven by the following three questions:
- Does TTN c.49648+2del mutation result in dilated cardiomyopathy?
- Were there any additional genetic or environmental factors that contributed to her disease phenotype?
- What are the mechanisms by which TTN c.49648+2del mutation results in cardiomyocyte dysfunction?
Background and Diagnosis
DCM is when ventricles in the heart become thin, stretched out, and fail to pump. There are many known causes of DCM, including stress-induced, ischemic, infectious, toxin-mediated, autoimmune, infiltrative, endocrinologic, nutritional, and genetic etiologies. DCM is classified as idiopathic when all identifiable causes (other than genetic etiologies) have been excluded. Genetic causes of DCM, termed 'familial' if present within families, make up ~30% of all cases (Neth Heart J). Familial DCM specifically is defined as idiopathic DCM that is present in two or more family members.
Dozens of genetic alterations are known to be associated with familial DCM. These include genes involved in a wide variety of cardiomyocyte structures and functions, including sarcomeres (TTN, MYH6, MYH7, TNNT2, TPM1, and MYPN), RNA splicing (RBM20), nuclear lamina integrity (LMNA), desmosome function (DSP), and ion channel proteins (SCN5A). TTN mutations comprise the majority (31%) of mutations underlying familial dilated cardiomyopathy (Sci Rep). These mutations are often autosomal dominant but can also arise de novo within individuals. TTN encodes titin protein, a large (3-4 MDa) and highly abundant protein that spans half of the sarcomere within myofilaments of striated muscle and serves as a 'biological spring' connecting the Z-disk to the M-line with functions in scaffolding and signaling (Pflugers Arch). TTN mutations most often result in truncated titin variants that are believed to lead to either haploinsufficiency or dominant negative effects within cardiomyocytes.
The heterozygous TTN c.49648+2 del mutation identified in our patient was previously reported in two families with DCM (N Engl J Med). This mutation results in the aberrant exclusion of exon 214 from the mRNA, which then encodes a truncated form of titin because of a frameshift and premature stop codon. This truncated form of titin lacks almost the entire "A-band" domain of titin that binds to myosin in the sarcomere.
The mechanisms by which heterozygous mutations resulting in truncated titin can cause DCM remains incompletely understood and are actively being studied. Using models where induced pluripotent stem cells (iPSCs) can be differentiated into cardiomyocytes, it has been shown that these mutations affect cardiac myocyte organization, contractile function, and response to stressful stimuli (Science). Whether this is mediated by haploinsufficiency (loss of wild-type titin) and/or a dominant negative function of truncated titin (so-called 'poison peptides') remains unclear. Truncated titin protein has been shown to be present in the cardiac tissue of most patients with mutations encoding truncated titin (Sci Transl). Truncated titin has not been shown to be incorporated into functional cardiac sarcomeres but has been shown to exist in intracellular aggregates in cardiac myocytes. Evidence suggests that aberrant protein quality control is a feature of DCM caused by truncated titin mutations (Sci Transl).
Research investigating the molecular mechanisms by which truncated titin mutations cause DCM has included a variety of different pathogenic mutations. To our knowledge, however, there are no published peer-reviewed studies at this time that have specifically investigated how TTN c.49648+2 del mutation affects cardiac myocyte function. Given this context, we hypothesize that our patient's TTN c.49648+2 del mutation is the major driver of her DCM. Furthermore, we hypothesize that additional genetic or environmental hits contributed to her relatively early-onset and more aggressive disease phenotype.
Summary and Future Steps
In summary, we present the case of a young woman presenting with acute decompensated heart failure in the setting of familial DCM, found to have a pathogenic truncating mutation in the titin gene. We propose that our patient's splice-site titin mutation results in truncated titin, which functions in both a haploinsufficiency and a dominant negative fashion to impair appropriate sarcomere organization and function and ultimately result in cardiomyocyte dysfunction and DCM.
Herein, we propose a series of genomic and functional assays to enable the characterization of the TTN c.49648+2del mutation and evaluate its impact on DCM alone and in combination with other putative DCM-inducing insults. Specifically, we propose:
- Whole genome sequencing of our patient, her sister, and their family members (Figure 1) to determine the prevalence of the TTN c.49648+2del mutation in her family and glean insights into disease penetrance. Additionally, this would enable us to appreciate other genetic variants present in her family that may modify disease severity.
- Develop an in vitro model of her TTN c.49648+2del mutation by generating patient-derived iPSCs from her available tissue biopsy (Figure 2). Through this model we aim to address the following questions:
- Does TTN c.49648+2del mutation encode a truncated form of titin?
- Does mutant TTN protein exhibit aberrant localization within the cardiomyocyte? Similarly, are there significant differences in sarcomere length and organization when TTN is mutated in the context of her genetic background?
- Does mutant TTN protein result in aberrant contractility?
- Does the expression of mutant TTN protein result in broad transcriptional rewiring that can have overarching impacts on cardiomyocyte function and stress responses?
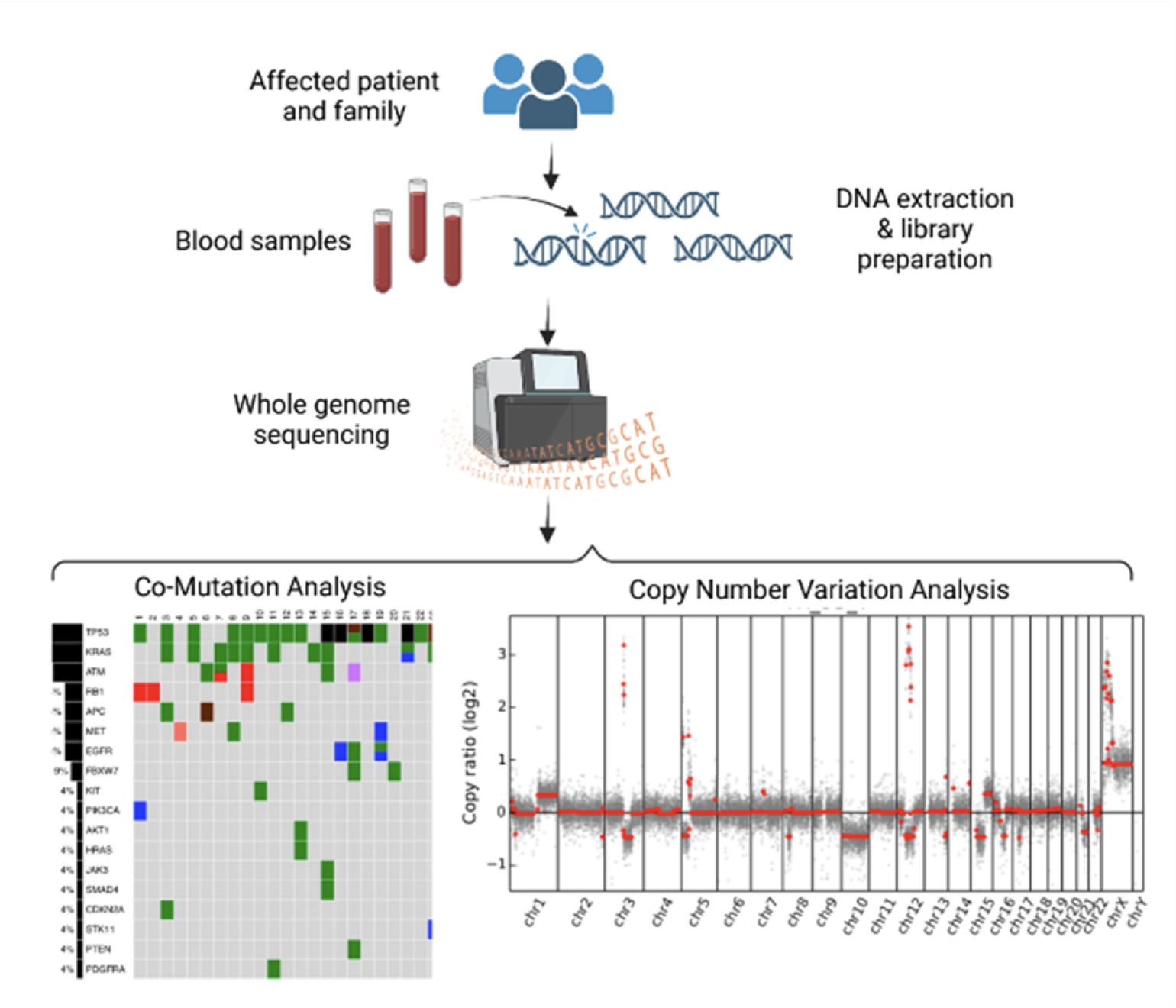
Figure 1
Proposed genomic sequencing of patient and family members, including co-mutation and copy number variation analyses, to determine the prevalence of the TTN mutation in the family and to identify other genetic variants that may contribute to disease phenotype.
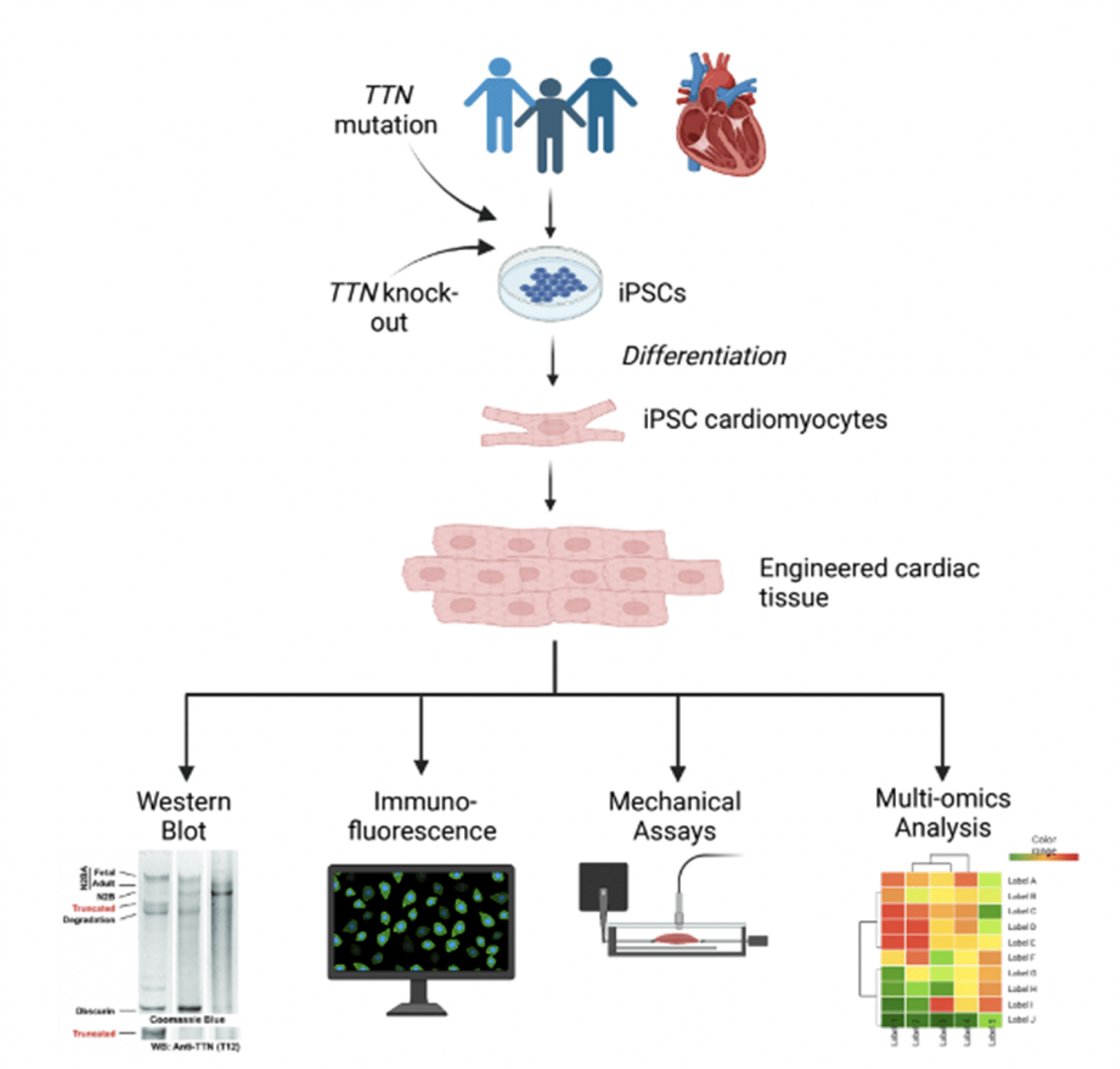
Figure 2
Proposed in vitro experimental approaches using iPSCs to determine the expression, localization, function, and transcriptional implications of the protein encoded by the TTN c.49648+2del mutation.
Learn more about the Pathways Consult Service at Mass General
Learn about research in the Department of Medicine