Antibodies in Severe COVID-19
The FLARE Four
- A subset of individuals with severe COVID-19 exhibit high levels of inflammatory cytokines, suggesting that an overexuberant immune response may play a key role in pathogenesis
- Antibody-dependent enhancement (ADE) is an immune phenomenon best described in dengue virus infection, where antibodies elicited by infection with one viral serotype paradoxically enhance both infection and host inflammatory response upon exposure to a newly infecting serotype
- ADE may occur during SARS-CoV-2 infection, leading to enhanced viral replication within immune cells while simultaneously promoting inflammation
- However, ADE is unlikely to be the sole determinant of severe COVID-19. For example, children exhibit ADE in other contexts but rarely develop severe COVID-19. Future studies will be needed to define any immune-mediated mechanisms driving severe SARS-CoV-2 infection
Many people are saying...severe COVID-19 is caused by the immune system. How could that be?
Subscribe to the latest updates from FLARE Advances in Motion
Introduction
SARS-CoV-2 infection in humans leads to remarkably variable presentations, ranging from asymptomatic viral shedding to acute respiratory distress syndrome and multi-organ failure (Cao 2020; Bai et al. 2020). Severe COVID-19 is associated with a failure to clear SARS-CoV-2 despite high levels of inflammatory cytokines, suggesting a dysfunctional immune response may drive pathogenesis (Liu et al. 2020). Recently, antibody-dependent enhancement (ADE) has been suggested as an immune mechanism promoting severe COVID-19 (Tetro 2020). In this FLARE, we will discuss the immunology of ADE and whether it may play a role in the variable host response to SARS-CoV-2 infection.
First, Let's Define Some Terms for Non-Immunologists
Antigen: a molecule that induces antibody generation such that the resulting antibody directly binds the inducing molecule.
Fc region: the “tail end” of an antibody, which does not bind to antigen, but interacts with immune cell surface receptors (termed Fc receptors) and some components of the complement system.
(The term Fc is an abbreviation for “fragment crystallizable” and stems from early studies whereby the tail end of the antibody molecule could be crystallized for protein structure experiments.)
Neutralization: the ability of antibodies to block pathogens from infecting target cells. Not all binding antibodies are neutralizing, either because they bind to portions of the virus not essential for entry or because they are not present at adequately high concentration (“sub-neutralizing”).
Serotype: a group within a species of microorganisms that induce distinct antibody responses due to differing surface antigens.
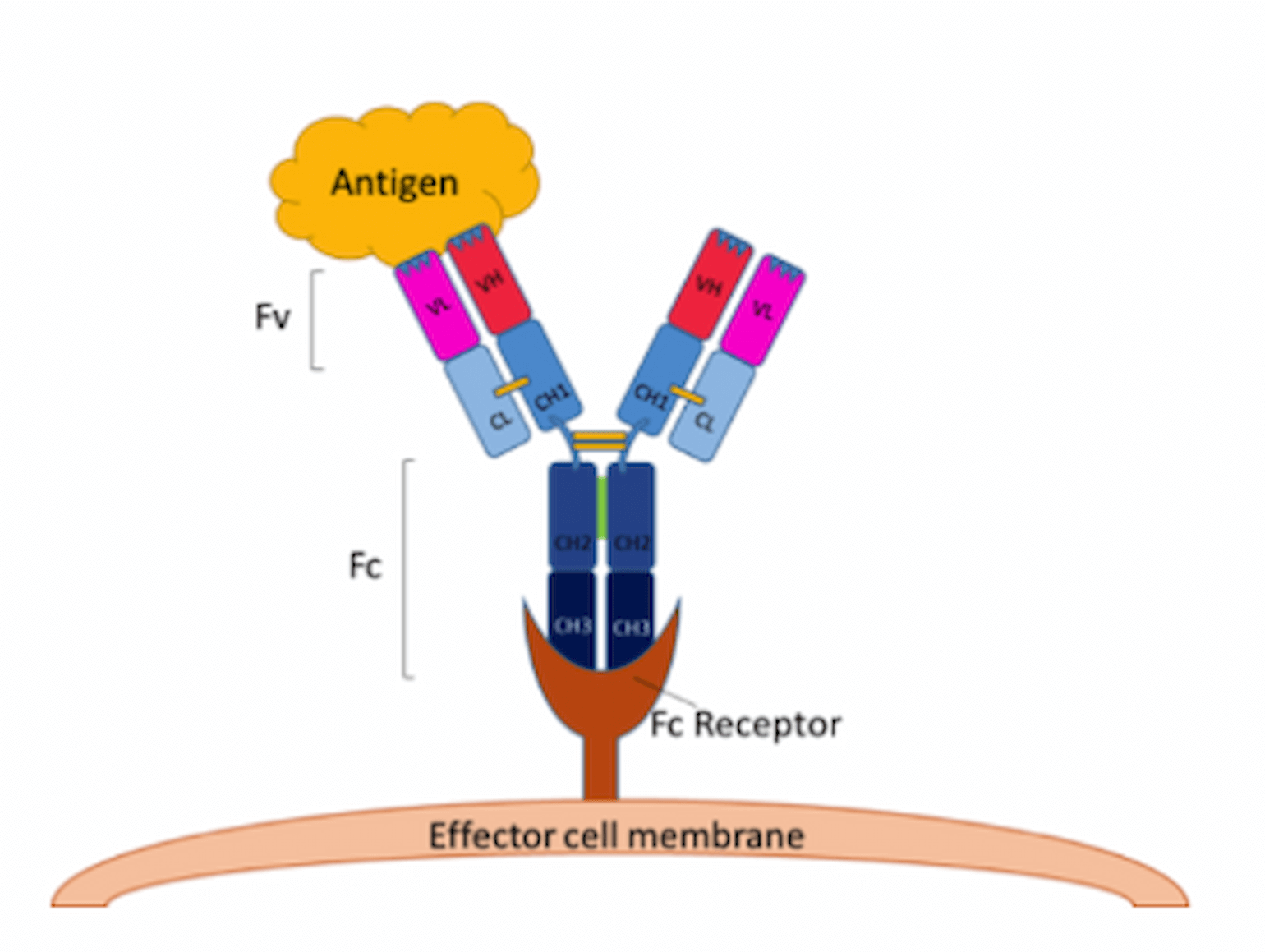
Figure 1
Figure from Bite-Size Bio.
How Do Antibodies Combat Viruses?
Antibodies play an important role in host defense against viral infections by binding viral surface antigens (Kulkarni 2020), thereby preventing entry and infectivity. Efficient anti-viral activity of antibodies also requires Fc-mediated mechanisms, which include complement-mediated viral particle lysis, phagocytosis, and killing of infected cells expressing surface viral proteins (Newton et al. 2016). Ultimately, effective neutralization is determined by both the affinity of the antibody and by accessibility of the recognized viral surface antigen (Dowd et al. 2011).
What is Antibody-Dependent Enhancement?
Viruses can acquire mutations that alter immunogenic surface antigens, leading to distinct viral serotypes. Consequently, the development of neutralizing antibodies to one viral serotype may not protect the host from a second serotype, even if antibody-binding to the second serotype occurs. Moreover, sub-neutralizing antibody levels can be detrimental to the host during viral infection. Antibody-dependent enhancement (ADE) describes the phenomenon where antibodies elicited by infection with one viral serotype not only fail to prevent infection by a newly infecting serotype, but actually enhance infection and the resulting inflammatory response. ADE occurs when antibody-mediated uptake of virus into immune cells that express Fc receptors (particularly macrophages and other monocytes) drives enhanced viral replication and cytokine production (Feinberg and Ahmed 2017).
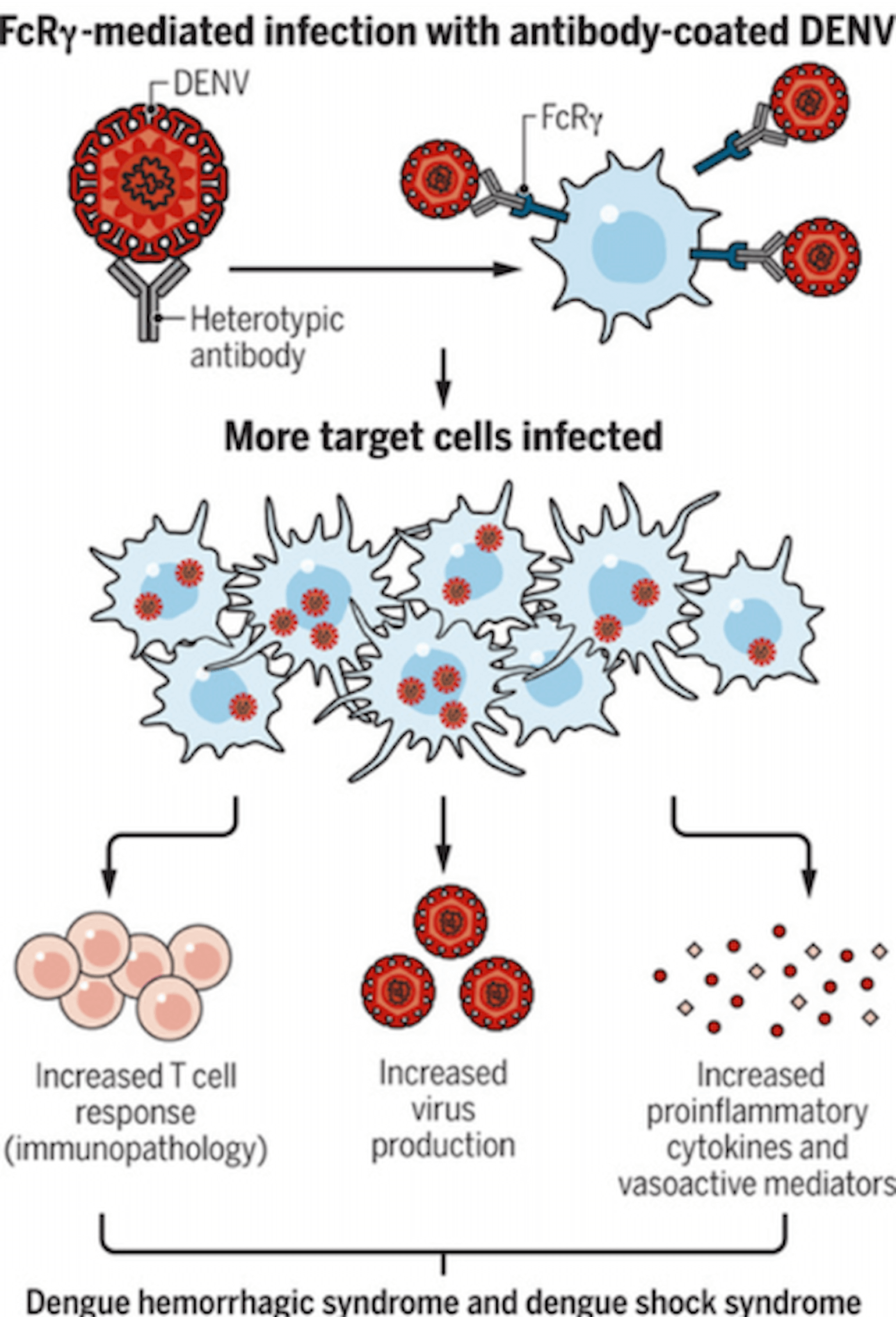
Figure 2
ADE, in combination with increased T cell responses and inflammatory cytokines, can increase severity of clinical symptoms (Feinberg and Ahmed 2017).
Importantly, the titer of antibodies is critical to the development of ADE, such that very low quantities do not cause significant enhancement while high titers may effectively neutralize virus even if the binding affinity is relatively weak (Feinberg and Ahmed 2017; Katzelnick et al. 2017). When ADE does occur, it can broaden viral tropism (i.e. which cells the virus can infect) and lead to more efficient viral entry than cognate receptor-mediated endocytosis. Along with enhancing viral replication, ADE can induce an over-exuberant innate and subsequently adaptive immune response, leading to immunopathology.
Antibody-Dependent Enhancement in Dengue Virus Infection
The ADE hypothesis originated in studies on dengue virus (DENV), a mosquito-borne tropical flavivirus (Wilder-Smith et al. 2019). Most individuals with DENV infection are asymptomatic or minimally symptomatic, and an additional ~25% of patients experience a self-limited febrile illness (Wilder-Smith et al. 2019; St. John and Rathore 2019). A small proportion of patients exhibit a profound inflammatory syndrome termed dengue hemorrhagic fever/dengue shock syndrome (DHF/DSS), which includes systemic vascular leak, coagulation abnormalities, and hemorrhage with multi-organ dysfunction. Interestingly, patients experiencing a second DENV infection with a distinct serotype are at increased risk for DHF/DSS, leading to the hypothesis that prior exposure to a different DENV serotype might somehow be responsible (Kulkarni 2020).
Indeed, multiple observations support the ADE hypothesis in DHF/DSS in vivo (Elong Ngono and Shresta 2018). First, non-human primates infected with DENV experience worsened viremia if they have previously been infected with a distinct DENV serotype (Halstead et al. 1973). In addition, monkeys that received DENV immune human sera or a monoclonal anti-DENV antibody exhibit higher viral loads than control monkeys (Halstead 1979; Goncalvez et al. 2007). Lastly, in humans, DHF/DSS does not seem to occur during primary DENV infection except in infants, but in all of these cases the mothers have possessed DENV-specific antibodies, suggesting ADE via passively acquired antibody (Halstead 1988). While these observations support the ADE hypothesis in dengue viral infection, notably, only a subset of individuals infected with a second DENV serotype develop DHF/DSS (Wilder-Smith et al. 2019). As a result, while ADE is likely necessary for DHF/DSS, it does not seem sufficient. There are almost certainly additional host factors that determine whether severe disease develops upon infection with a second DENV serotype (Wilder-Smith et al. 2019).
Is There Evidence of ADE During Coronavirus Infection?
The concept that ADE might play a role during coronavirus infection has been considered since the SARS outbreak in 2003 (Tetro 2020). Coronaviruses represent a broad family of RNA viruses that commonly infect humans (typically causing mild upper respiratory tract infections), so many individuals likely harbor anti-coronavirus antibodies (Channappanavar and Perlman 2017). Yang and colleagues first proposed that prior infection with non-SARS coronavirus could promote SARS (Yang et al. 2005). Human coronavirus 229E (CoV-229E), responsible for the common cold, has been proposed as a priming serotype leading to ADE upon SARS-CoV infection (Ho et al. 2005). Subsequent studies suggested that sub-neutralizing concentration of antibodies binding to the spike protein of SARS-CoV (elicited by a SARS-CoV vaccine candidate), which mediates viral entry, were responsible for infection of Fc-bearing immune cells (Jaume et al. 2011; Yip et al. 2014).
Since the SARS-CoV-2 outbreak, there has been additional speculation that ADE is responsible for severe COVID-19 (Tetro 2020). While at this time there is no direct evidence for ADE in COVID-19, this mechanism would offer a potential explanation for the variable clinical presentations observed. Specifically, prior infection with a non-SARS coronavirus might result in a sub-neutralizing level of coronavirus antibodies, enhancing SARS-CoV-2 infection, and thereby leading to severe COVID-19.
What is Problematic About This Hypothesis in SARS-CoV-2 Infection?
While intriguing, there are limitations to using this model to explain the observed patterns of COVID-19 illness. In particular, if ADE is the dominant immune-mediated mechanism driving pathologic responses to SARS-CoV-2, it is far from clear why children would be generally spared from severe COVID-19 (please see April 7 FLARE). As outlined below, one can offer possible explanations, but none fully account for the observed relatively attenuated infection in children:
- Hypothesis: Children may have never been exposed to the requisite priming coronavirus
- Counterpoint: This is unlikely, however, as coronavirus infection in children is common (Talbot et al. 2009). Alternatively, this would require that the specific priming coronavirus has not infected humans in decades, which seems unlikely.
- Hypothesis: Children may not have coronavirus-specific antibody titers capable of inducing ADE
- Counterpoint: While it is well-documented that infants have relatively poor humoral immune responses to respiratory viral infections, passive transfer of maternal antibody should give some infants ADE-inducing, coronavirus-specific antibody titers (Heinonen et al. 2019). In addition, in contrast to the weak humoral immune response of infants, older children would presumably generate sufficient long-lived, coronavirus-specific antibodies, yet they still rarely develop severe COVID-19.
- Hypothesis: Unique features of antibody or Fc receptor biology or innate immunity in children precludes ADE during SARS-CoV-2 infection
- Counterpoint: However, children develop DHF/DSS, suggesting they are fully capable of executing ADE during viral infection.
Consequently, while ADE may be occurring in certain individuals infected with SARS-CoV-2, promoting both viral replication and an overly exuberant immune response, it seems less likely that sub-neutralizing levels of coronavirus-specific antibodies are the dominant immune-mediated mechanism driving severe COVID-19. Additional investigation into the immune response to SARS-CoV-2 is needed to delineate the mechanisms driving morbidity and mortality in COVID-19.
Conclusions
ADE is an interesting hypothesis which might explain the coexistence of increased markers of inflammation and delayed viral clearance reported in some patients with severe COVID-19. However, ADE alone likely does not fully explain the heterogeneity of host responses to SARS-CoV-2 infection. Specifically, children, who do exhibit ADE in other contexts, rarely develop severe COVID-19. As a result, if ADE contributes to COVID-19, there are likely additional host factors that dictate the severity of clinical presentation. Rigorous studies are needed to identify and characterize immune-mediated mechanisms which may play a role in some patients with severe COVID-19.
References
- Bai, Yan, Lingsheng Yao, TaoWei, Fei Tian, Dong-Yan Jin, Lijuan Chen, and MeiyunWang. 2020. “Presumed Asymptomatic Carrier Transmission of COVID-19.” Journal of the American Medical Association 323 (14): 1406–7.
- Cao, Xuetao. 2020. “COVID-19: Immunopathology and Its Implications for Therapy.” Nature Reviews Immunology 2019. https://doi.org/10.1038/s41577-020-0308-3.
- Channappanavar, Rudragouda, and Stanley Perlman. 2017. “Pathogenic Human Coronavirus Infections: Causes and Consequences of Cytokine Storm and Immunopathology.” Seminars in Immunopathology 39 (5): 529–39. https://doi.org/10.1007/s00281-017-0629-x.
- Dowd KA, Jost CA, Durbin AP, Whitehead SS, Pierson TC. 2011. “A Dynamic Landscape for Antibody Binding Modulates Antibody-Mediated Neutralization of West Nile Virus.” PLoS Pathogens 7 (6): e1002111.
- Elong Ngono, Annie, and Sujan Shresta. 2018. “Immune Response to Dengue and Zika.” Annual Review of Immunology 36 (1): 279–308. https://doi.org/10.1146/annurev-immunol-042617-053142.
- Feinberg, Mark B., and Rafi Ahmed. 2017. “Advancing Dengue Vaccine Development.” Science 358 (6365): 865–66. https://doi.org/10.1126/science.aaq0215.
- Goncalvez, Ana P., Ronald E. Engle, Marisa St. Claire, Robert H. Purcell, and Ching Juh Lai. 2007. “Monoclonal Antibody-Mediated Enhancement of Dengue Virus Infection in Vitro and in Vivo and Strategies for Prevention.” Proceedings of the National Academy of Sciences of the United States of America 104 (22): 9422–27. https://doi.org/10.1073/pnas.0703498104.
- Halstead, Scott B; Shotwell, Henry; Casals, Jordi. 1973. “Studies on the Pathogenesis of Dengue Infection in Monkeys . II . Clinical Laboratory Responses to Heterologous Infection.” The Journal of Infectious Diseases 128 (1): 15–22.
- Halstead, Scott B. 1979. “In Vivo Enhancement of Dengue Virus Infection in Rhesus Monkeys by Passively Transferred Antibody.” The Journal of Infectious Diseases 140 (4): 527–33. 1988. “Pathogenesis of Dengue: Challenges to Molecular Biology.” Science 239 (4839): 476–81.
- Heinonen, Santtu, Rosa Rodriguez-Fernandez, Alejandro Diaz, Silvia Oliva Rodriguez-Pastor, Octavio Ramilo, and Asuncion Mejias. 2019. “Infant Immune Response to Respiratory Viral Infections.” Immunology and Allergy Clinics of North America 39 (3): 361–76. https://doi.org/10.1016/j.iac.2019.03.005.
- Ho, Mei Shang, Wei Ju Chen, Hour Young Chen, Szu Fong Lin, Win Chin Wang, Jiali Di, Yen Ta Lu, et al. 2005. “Neutralizing Antibody Response and SARS Severity.” Emerging Infectious Diseases 11 (11): 1730–37. https://doi.org/10.3201/eid1111.040659.
- Jaume, M., M. S. Yip, C. Y. Cheung, H. L. Leung, P. H. Li, F. Kien, I. Dutry, et al. 2011. “Anti-Severe Acute Respiratory Syndrome Coronavirus Spike Antibodies Trigger Infection of Human Immune Cells via a PH- and Cysteine Protease-Independent Fc R Pathway.” Journal of Virology 85 (20): 10582–97. https://doi.org/10.1128/jvi.00671-11.
- John, Ashley L. St., and Abhay P.S. Rathore. 2019. “Adaptive Immune Responses to Primary and Secondary Dengue Virus Infections.” Nature Reviews Immunology 19 (4): 218–30. https://doi.org/10.1038/s41577-019-0123-x.
- Katzelnick, Leah C., Lionel Gresh, M. Elizabeth Halloran, Juan Carlos Mercado, Guillermina Kuan, Aubree Gordon, Angel Balmaseda, and Eva Harris. 2017. “Antibody-Dependent Enhancement of Severe Dengue Disease in Humans.” Science 358 (6365): 929–32. https://doi.org/10.1126/science.aan6836.
- Kulkarni, Ruta. 2020. Antibody-Dependent Enhancement of Viral Infections. Dynamics of Immune Activation in Viral Diseases. https://doi.org/10.1007/978-981-15-1045-8.
- Liu, Yang, Li Meng Yan, Lagen Wan, Tian Xin Xiang, Aiping Le, Jia Ming Liu, Malik Peiris, Leo L.M. Poon, and Wei Zhang. 2020. “Viral Dynamics in Mild and Severe Cases of COVID-19.” The Lancet Infectious Diseases 2019 (20): 2019–20. https://doi.org/10.1016/S1473-3099(20)30232-2.
- Newton, Amy H., Amber Cardani, and Thomas J. Braciale. 2016. “The Host Immune Response in Respiratory Virus Infection: Balancing Virus Clearance and Immunopathology.” Seminars in Immunopathology 38 (4): 471–82. https://doi.org/10.1007/s00281-016-0558-0.
- Tetro, Jason A. 2020. “Is COVID-19 Receiving ADE from Other Coronaviruses?” Microbes and Infection 22 (2): 72–73. https://doi.org/10.1016/j.micinf.2020.02.006.
- Wilder-Smith, Annelies, Eng Eong Ooi, Olaf Horstick, and Bridget Wills. 2019. “Dengue.” The Lancet 393 (10169): 350–63. https://doi.org/10.1016/S0140-6736(18)32560-1.
- Yang, Zhi Yong, Heidi C. Werner, Wing Pui Kong, Kwanyee Leung, Elisabetta Traggiai, Antonio Lanzavecchia, and Gary J. Nabel. 2005. “Evasion of Antibody Neutralization in Emerging Severe Acute Respiratory Syndrome Coronaviruses.” Proceedings of the National Academy of Sciences of the United States of America 102 (3): 797–801. https://doi.org/10.1073/pnas.0409065102.
- Yip, Ming Shum, Nancy Hiu Lan Leung, Chung Yan Cheung, Ping Hung Li, Horace Hok Yeung Lee, Marc Daëron, Joseph Sriyal Malik Peiris, Roberto Bruzzone, and Martial Jaume. 2014. “Antibody-Dependent Infection of Human Macrophages by Severe Acute Respiratory Syndrome Coronavirus.” Virology Journal 11 (1): 1–11. https://doi.org/10.1186/1743-422X-11-82.
View all COVID-19 updates
Learn about research in the Division of Pulmonary and Critical Care Medicine