Pathways Case Report: Anti-HMGCR Statin-associated Myopathy
In This Case Study
- A 69-year-old man with a past medical history of a non-ST-segment elevation myocardial infarction (NSTEMI) with drug-eluting stent placement approximately three and a half years prior was admitted with back pain radiating to the shoulders and hip
- His recent history was notable for complaints of intermittent weakness in both legs and arms as well as difficulties swallowing over several months
- The Pathways Service in the Department of Medicine at Massachusetts General Hospital was consulted during the patient's admission
A 69-year-old man with a past medical history of a non-ST-segment elevation myocardial infarction (NSTEMI) with drug-eluting stent placement approximately three and a half years prior was admitted to the Massachusetts General Hospital with back pain radiating to the shoulders and hip. His recent history was notable for complaints of intermittent weakness in both legs and arms as well as difficulties swallowing over several months. At the time of these complaints (~three months before hospitalization), he had elevated liver chemistries and creatine phosphokinase (CPK), which is consistent with a diagnosis of statin myopathy. His presentation was initially thought to be secondary to the high dose atorvastatin that he began taking after his NSTEMI, which was discontinued at the time.
Subscribe to the latest updates from Cardiovascular Advances in Motion
Upon admission three months after stopping atorvastatin therapy, he had limited clinical improvement. He had continued musculoskeletal complaints and sustained CPK elevation that had incompletely resolved despite statin cessation. The inpatient rheumatology team was consulted and recommended measuring antibodies directed against HMG-CoA reductase (HMGCR, an enzyme in cholesterol synthesis), which were elevated in our patient, resulting in the diagnosis of statin-associated anti-HMGCR myopathy. The patient declined further diagnostic testing and treatment (including MRI, muscle biopsy, and immunosuppression with steroids or intravenous immunoglobulin), and was discharged with a stable pain management regimen for his paraspinal back pain.
The Pathways Service in the Department of Medicine at Massachusetts General Hospital was consulted during the patient's admission, and we were intrigued by the pathophysiology of this rare clinical entity, anti-HMGCR myopathy (seen in 2–3 per 100,000 person-years in patients taking statins1) and its clinical similarities with the much more common statin-induced myalgias (up to 30% of patients on statins2). From there, we developed a line of clinical and scientific inquiry focused on the following questions:
- What is the pathogenesis of anti-HMGCR myopathy?
- Is anti-HMGCR myopathy related to general statin myopathy?
Background and Diagnosis
Humans require cholesterol to maintain membrane fluidity and function as the precursor to crucial molecules such as bile acids and steroid hormones3. Cells obtain cholesterol through two mechanisms: 1) through an endogenous biosynthetic pathway starting with acetyl-CoA, and 2) through the receptor-mediated uptake of lipoproteins such as low-density lipoprotein (LDL). In the endoplasmic reticulum, the sterol-regulatory element binding protein (SREBP) pathway senses the amount of cellular cholesterol and, in the setting of low LDL (Figure 1A, top), upregulates genes encoding for biosynthesis of cholesterol (i.e., HMGCR) and the LDL receptor (LDLR), leading to higher production and uptake of cholesterol by the cell, respectively. Conversely, in the setting of high LDL, the cell will turn off the SREBP pathway and replete cellular cholesterol (Figure 1A, middle).
Statins, medications that are prescribed to millions of patients, exploit this system by blocking the rate-limiting enzyme in cholesterol synthesis (i.e., HMGCR), thereby activating the SREBP pathway despite normal cellular cholesterol levels (Figure 1A, bottom). This leads to decreased circulating plasma LDL and reduces the incidence of coronary heart disease4. Notably, statin treatment prevents cholesterol biosynthesis (Figure 1B) but also thwarts cells from producing other essential molecules such as isopentenyl adenosine (required for certain tRNAs) and farnesyl phosphate (used to make dolichol for glycosylation, vitamins A, E, and K, heme, ubiquinone for mitochondrial electron transport, and lipid anchors for proteins such as Ras)5.
Statin-related myopathies are separate clinical entities that present similarly to other myopathies (i.e., proximal muscle weakness and pain in the setting of statin use) but are separated based on CPK elevation. While the asymptomatic CPK elevation and myalgias can self-resolve, the cornerstone of treatment for statin myopathies and rhabdomyolysis is statin cessation. The order of increasing severity and decreasing prevalence are outlined in Table 1. For statin-associated autoimmune myopathy, MRI and muscle biopsy may be obtained if myonecrosis is suspected1. Statin discontinuation is not curative, and severe cases may require immunosuppressive therapy. Of note, the development of statin myopathy has predisposing risk factors related to the bioavailability of statins, including CYP3A4 inhibition6, and haplotypes in the liver statin exporter OATP1B1, encoded by SLC01B1. The development of immune-mediated necrotizing myopathy has the predisposing factor of the class II HLA allele HLA-DRB1*11:01 haplotype, which has an odds ratio of 25-507.
Table 1.
| Disease | Muscle symptoms | CPK Elevation (above upper limit of normal) |
anti-HMGCR antibody |
| Asymptomatic CPK elevation | No | < 4-fold | Negative |
| Statin-related myalgia | Yes | < 4-fold | Negative |
| Statin-related myopathy | Yes | 4- to 10-fold | Negative |
| Statin-related rhabdomyolysis | Yes | 10- to 50-fold | Negative |
| Statin-associated autoimmune myopathy | Yes | 10- to 50-fold | Positive |
Given the clinical similarities of the statin-related myopathies, the predisposing risk factors for statin-associated autoimmune myopathy relating to altered pharmacokinetics, and the 3.5-year latency period from statin therapy until the presentation of our patient, we hypothesized that there may be a stepwise continuum of the statin-related myopathies that culminates in anti-HMGCR myonecrosis, that is caused by on-target effects of statins.
Summary and Future Steps
To test our hypothesis, we propose three scientific lines of questioning to investigate statin-related myopathies, their relation to one another, and the cellular effects of statin therapies.
First, we suggest a longitudinal study defining the natural history of anti-HMGCR mediated disease starting with a large population of patients taking statins, with a goal to prospectively identify serial measurements of CPK, anti-HMGCR antibodies, and muscle-specific markers of apoptosis and regeneration.
Second, we propose a study to identify the mevalonate product whose deficiency causes statin myopathy. A recent study8 revealed that treatment with mevalonolactone (the lactone form of mevalonate, the product of HMGCR) improved muscle function in both patients with a genetic limb-girdle muscular dystrophy caused by partial loss of function mutations in HMGCR as well as in a mouse model of statin myopathy. Plasma cholesterol-containing lipoprotein levels (i.e., LDL) were not affected by treatment, indicating that lack of one of the derivatives of isopentenyl or farnesyl pyrophosphate is necessary for normal muscle function. We propose systematically adding these metabolites back to the mouse model of statin myopathy as well as a muscle cell line (C2C12 myotubes) bearing a CRISPR-generated knock-in mutation in HMGCR to see whether we can rescue the myopathy phenotype and markers of inflammation or cell death resulting from statin treatment.
Lastly, we recommend an experiment to isolate autoreactive lymphocytes from patients with anti-HMGCR antibodies and determine the difference in effects on lipid metabolism in the presence of statins when compared to control T-lymphocytes.
The findings from these studies will not only help better understand statin myopathy and the development of statin-associated autoimmune myopathy but could lead to preventive strategies to increase statin adherence and, therefore, improve cardiovascular health by subverting clinically significant muscular side effects.
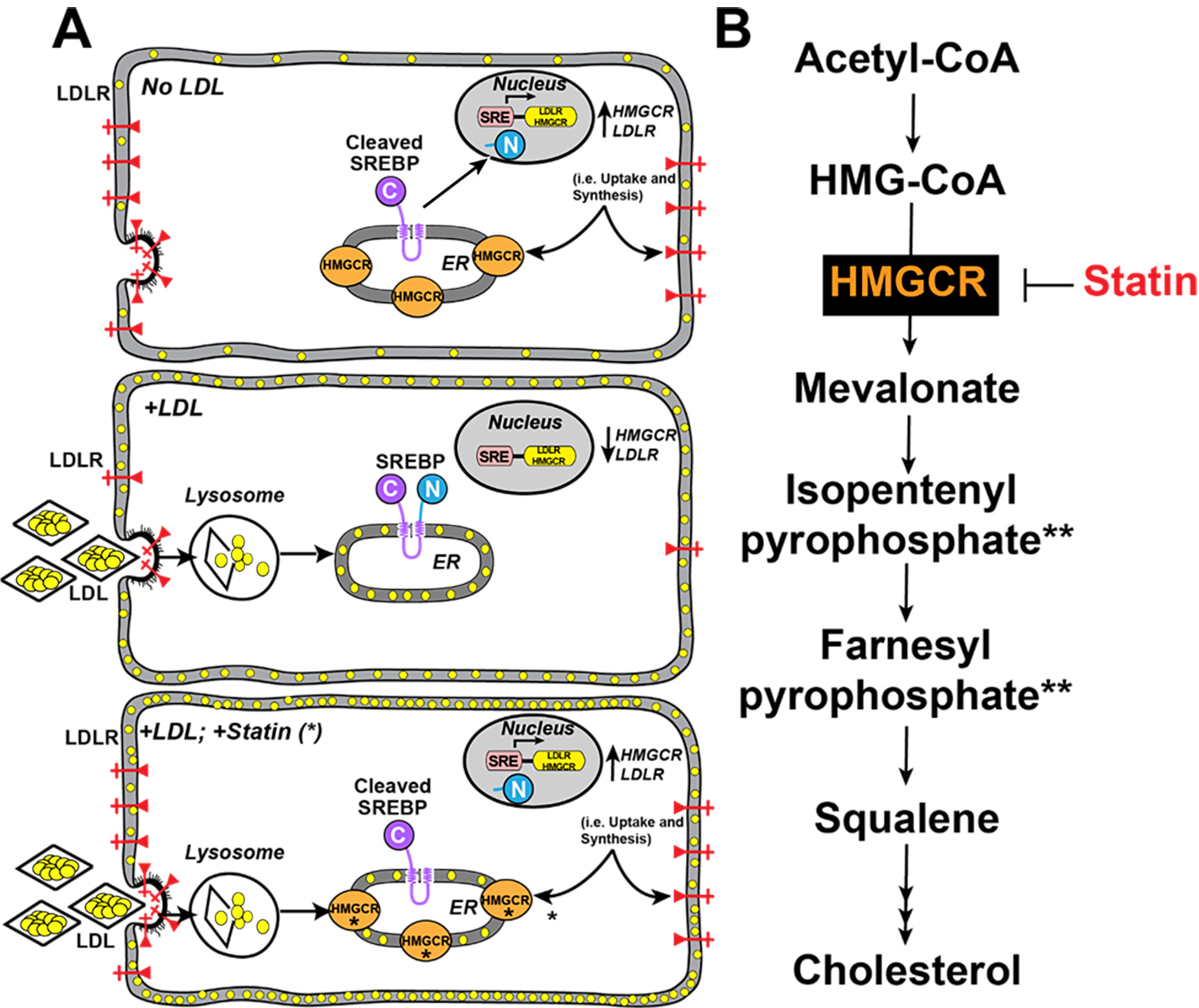
Figure 1
Figure 1. Regulation of Cellular Cholesterol. (A) Top, the production of cholesterol through biosynthesis as well as LDL receptors is controlled by a membrane-bound transcription factor in the ER called SREBP. When the cells are depleted of cholesterol (yellow circles), SREBP is processed by proteases to release a soluble domain that travels to the nucleus and activates transcription of the cholesterol biosynthesis genes (e.g., HMGCR) and the LDL receptor (LDLR) gene. Middle, when cells are replete with cholesterol, the LDL binds to LDL receptors that incorporate the LDL into vesicles that deliver the LDL to lysosomes where the LDL is degraded. LDL cholesterol is liberated and the cholesterol is delivered to the plasma membrane and the ER, where it blocks the proteolytic processing of SREBP. As a result, the transcription of HMGCR and LDLR declines. Bottom, statins (*) inhibit HMGCR, so despite adequate LDL uptake, SREBP continues to upregulate HMGCR and LDLR to remove circulating plasma lipoproteins. (B) Cholesterol Biosynthesis. HMGCR is inhibited by statin. (**) denoted mevalonate-derived molecules whose deficiency may cause statin myopathy.
References
- Mammen AL. (2016) Statin-Associated Autoimmune Myopathy. N Engl J Med. 374(7):664-9. doi: 10.1056/NEJMra1515161.
- Turner RM, Pirmohamed M. (2019). Statin-Related Myotoxicity: A Comprehensive Review of Pharmacokinetic, Pharmacogenomic and Muscle Components. J Clin Med. 9(1):22. doi: 10.3390/jcm9010022.
- Goldstein JL, Brown MS. (2015) A century of cholesterol and coronaries: from plaques to genes to statins.Cell.161(1):161-172. doi: 10.1016/j.cell.2015.01.036.
- Scandinavian Simvastatin Survival Study Group (1994). Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease. Lancet. 344(8934):1383-9.
- Lodish H, Berk A, Kaiser CA et al. (2021). Molecular Cell Biology 9th Edition. New York:
- Vinci P. et al. (2021). Statin-Associated Myopathy: Emphasis on Mechanisms and Targeted Therapy. Int J Mol Sci. 22(21):11687. doi: 10.3390/ijms222111687.
- Mammen AL, Gaudet D, Brisson D, Christopher-Stine L, Lloyd TE, Leffell MS, Zachary AA. (2012). Increased frequency of DRB1*11:01 in anti-hydroxymethylglutaryl-coenzyme A reductase-associated autoimmune myopathy. Arthritis Care Res (64(8):1233-7. doi: 10.1002/acr.21671.
- Yogev Y, et al. (2023). Limb girdle muscular disease caused by HMGCR mutation and statin myopathy treatable with mevalonolactone. PNAS .120, e2217831120.